The evolution and conservation of lncRNA sequences has been a topic of intense study and debate since the vast numbers of lncRNA transcripts were discovered (3,6). Determining if lncRNAs as a group are conserved and evolve over time is important because it would provide evidence that they are functionally important to organisms and not merely the product of transcriptional noise.
The function of lncRNAs have been questioned ever since their discovery through cDNA library sequencing and microarray tiling arrays. Both these techniques quantify the presence of these transcripts, but do not provide any information about their activities in the cell. Because of this uncertainty, one hypothesis for the existence of lncRNA is transcriptional “noise” due to low fidelity binding of RNA polymerase to randomly occurring weak promoter sequences in the genome (R1,R2,R3). This hypothesis implies that it is more energy efficient for the cell to allow some random transcription to occur than it would be to downregulate nonspecific RNA polymerase binding and transcription. One piece of evidence for this hypothesis is that although the quantity of individual lncRNA transcripts is much greater than mRNA transcripts, individual lncRNA transcripts are transcribed at much lower levels than individual mRNAs (7).
Another piece of evidence that may support the transcriptional noise hypothesis is the generally low conservation of lncRNA sequences between species (R2,6). There have been many studies that have calculated the rates of sequence change in lncRNAs over time. The general strategy used to calculate these rates is to compare the number of nucleotide changes in the sequence in question to the number of changes in sequences of the same size that are under not under selective pressure, such as transposable elements (R3). Although the results of the studies vary depending on which lncRNA sequences are studied and the sequences that are used as controls, a recent study that screened for lncRNAs with relatively high degrees of sequence conservation identified a group of ~3000 lncRNAs (8). However, it is important to note that even this group of lncRNAs had very low sequence conservation as compared to protein coding genes; when the transposable element control group was normalized to a 100% sequence change rate, protein coding genes were calculated to have a 10% change rate, whereas lncRNA sequences had a 95% change rate. Nevertheless, studies such as this one mayidentify lncRNAs that have a higher probability of functional importance as subjects for further study.
Another explanation for the low sequence conservation of lncRNAs is that they might not require very much nucleotide sequence conservation to maintain their functionality. Protein coding genes are under very intense selection restraints due to their need to maintain the correct amino acid coding and open reading frame. In contrast, RNA molecules have less rigid sequence requirements to maintain their secondary structures and may only need to maintain short stretches of conserved sequence to keep normal function(R1,R2,R3). An example of this flexibility is seen in the lncRNA Xist. Xist silences one of the two X chromosomes in all eutherian females to achieve proper X-linked gene dosage. Despite this well-defined and essential role, Xist shows very little sequence conservation throughout the eutherian lineage, demonstrating the fact that a high degree of sequence conservation is not an essential requirement for lncRNA functionality (9).
Rapid evolution of lncRNA sequences has also been hypothesized to be a reason for the low level of lncRNA sequence conservation. Due to the flexible relationship between lncRNA sequence and function, lncRNAs may be more plastic and thus more amenable to evolutionary change than protein coding genes. As the list of organisms with fully sequenced genomes has grown a striking pattern has emerged. Although the numbers of genes do not increase with organism complexity as it was originally assumed they would, the numbers of lncRNAs increase dramatically (5,4). This correlation between lncRNA expansion and organism complexity is very suggestive and implies lncRNAs may be an important factor in evolutionary development. On a shorter evolutionary time scale, it has been shown that changes in lncRNA sequences constitute half of all the genetic differences between the human and chimpanzee genomes (10). However, it has yet to be determined if these changes are causative of the differences between humans and chimpanzees or if they are the result of genetic drift.
In contrast to the lack sequence conservation in the lncRNA sequences themselves, the promoters of lncRNAs show very high sequence conservation. In fact, a recent study in mice calculated the sequence conservation of lncRNA promoters to be higher than the sequence conservation of protein coding gene promoters (4). Transcription factors have also been shown to bind to the promoter regions of lncRNAs (R3). These data indicate that although lncRNA sequences might not be highly conserved, the level of their transcription is.
Origins of lncRNAs (R3):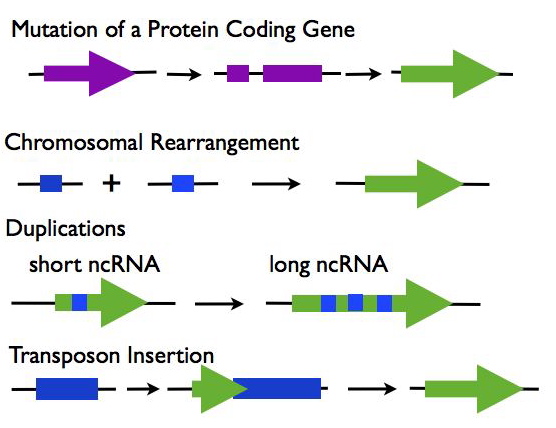
- Mutations in a Protein Coding Gene: A protein-coding gene may under go mutations such as a frame shift that disrupts its open reading frame while maintaining the expression of the RNA transcript.
- Chromosomal Rearrangement: Two separate sequences are joined which together create an expressed noncoding sequence.
- Duplications: Duplications in a noncoding RNA sequence cause repeats, increasing the length of the transcript.
- Transposable Element Insertion: A transposable element containing a transcriptional start site is inserted into the genome and creates a functional, but noncoding RNA sequence.
LncRNAs are a heterogenous group of transcripts with no defined set of parter RNAs or proteins, please refer to the Mechanism of Action section above for some examples of lncRNA interactions with partner molecules.
LncRNAs are a heterogenous group of molecules that regulate gene expression in many ways (R2,R3).
Regulation of Chromatin Structure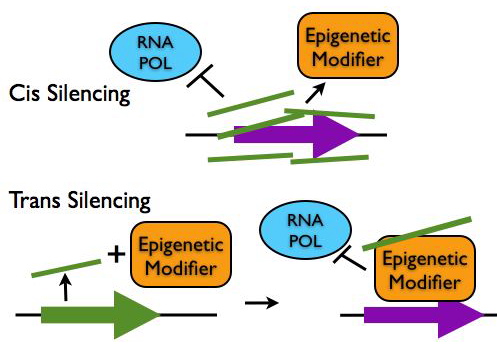
- Epigenetic Silencing in cis: LncRNA transcripts such as Xist and Air coat gene clusters and silence their expression by making them inaccessible to transcription machinery. These lncRNAs can also recruit chromatin remodeling proteins to epigenetically mark the region for heritable gene silencing.
- Epigentic Silencing in trans: LncRNAs such as HOTAIR can interact with chromatin modifying proteins to epigenetically silence genes at another locus.
Gene Regulation via lncRNA transcription![]()
- Activation via Transcription: The act of lncRNA transcription itself has been shown to open the chromatin structure of a genetic locus to permit access of transcription machinery to other protein coding genes. Transcription of lncRNAs UAS1 and UAS2 have been shown to activate the expression of the fbp1 gene in this way.
- Repression via Transcription: Transcription of lncRNAs near to protein coding loci can also repress gene transcription because the presence of the transcription machinery on the lncRNA gene locus physically prevents transcription machinery from binding to the protein coding gene. Transcription of the lncRNA SRG1 has been shown to inhibit transcription of the overlapping SER3 gene through this mechanism.
Transcription Regulation in cis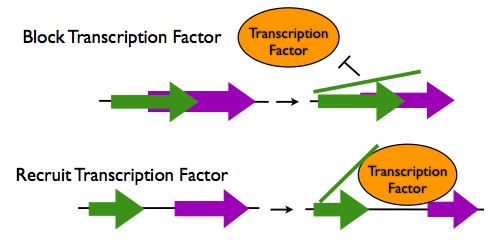
- Occlusion of a Transcription Factor Binding Site: If a lncRNA sequence overlaps with a transcription factor binding site, the lncRNA transcript can hybridize to this site to prevent a transcription factor from binding. One lncRNA binds to both the promoter of the DHFR gene and the transcription factor TFIIB to prevent transcription.
- Recruitment of Transcription Factors: When an lncRNA sequence is located near to transcription factor binding site, the lncRNA transcript can hybridize to both that location and to a transcription factor protein, thus enhancing the binding of the transcription factor. An example of this is the recruitment of the transcription factor DLX2 to the Dlx6 gene by the lncRNA Evf2.
Transcription Regulation in trans: ![]()
- Activation of Transcription Factors: LncRNA transcripts can activate transcription factors via allosteric interactions, such as the activation of the Dlx5/6 enhancer by the lncRNA Evf2.
- Transport of Transcription Factors: LncRNA transcripts can alter the trafficking of transcription factors in the cell, either enhancing their access to their binding sites or preventing it, as in the case of the lncRNA NRON preventing the transcription factor NFAT from entering the nucleus.
- Interactions with Accessory Proteins: LncRNA transcripts can bind to accessory proteins to activate them allosterically, or induce their oligomerization and activation as in the case of lncRNA-induced trimerization of HSF1 proteins in response to heat shock.
Post Transcriptional Regulation![]()
- Regulation of mRNAs: Many of the lncRNAs that are antisense to protein coding genes may function in regulating the splicing, editing, transport, translation, and degradation of their corresponding mRNA transcripts.
- Gene Silencing: Many lncRNAs may be processed into short ncRNAs such as siRNAs that can downregulate gene expression by degrading the mRNA transcripts. An example of this type of lncRNA is H19 which is processed into the microRNA miR-675 (R3).
Although many lncRNAs are known to be transcribed, very few of these have experimentally-defined functions. However, based on the lncRNAs that have been analyzed thus far, lncRNAs appear to be able to regulate gene expression through a diverse group of mechanisms.
R1) Louro R, Smirnova AS, Verjovski-Almeida S. Long intronic noncoding RNA transcription: Expression noise or expression choice? 2009. Genomics 93: 291-298. PMID
R2) Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. 2009. Nat Rev Genetics 10: 155-159. PMID
R3) Ponting CP, Oliver PL, Reik W. Evolution and Functions of Long Noncoding RNAs. 2009. Cell 136: 629-641. PMID:
1) Brannan CI, et al. The product of the H19 gene may function as an RNA. 1990. Mol Cell Bio 10 (1): 28-36.
2) Brockdorff N, et al. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. 1992. Cell. 71(3): 515-526.
3) Okazaki Y, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. 2002. Nature 420: 563-573.
4) Carninci P, et al. The transcriptional landscape of the mammalian genome. 2005. Science 309 (5740): 1559-63.
5) Huttenhofer A, Schattner P, Polacek N. Non-coding RNAs: hope or hype? 2005. Trends in Genetics 21: 289-297.
6) Wang J, et al. Neutral Evolution of ‘non-coding’ complementary DNAs. 2004. Nature 431:757.
7) Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 15: R17-R29.
8) Ponjavic J, et al. Functionality or Transcriptional Noise? Evidence for selection within long noncoding RNAs. 2007. Genome Res 17: 556-565.
9) Pang KC, Frith M Mattick JS. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. 2006. Trends Genet 22: 1-5
10) Khaitovitch P et al. Functionality of intergenic transcription: an evolutionary comparison. 2006. PLoS Genet. 2: e171.
11) Mercer TR, et al. Specific Expression of long noncoding RNAs in the mouse brain. 2008. Proc Nat Acad Sci USA 105: 716-721.
12) Amaral PP, Mattick JS. Noncoding RNA in Development. 2008. Mamm Genome 19: 454-492
13) Babak T, Blencowe BJ, Hughes TR. Considerations in the identification of functional RNA structural elements in genomic alignments. 2007. BMC Bioinformatics. 8:33.
14) Mallardo et al. Non-protein coding RNA biomarkers and differential expression in cancers: a review. 2008. J Clin Cancer Res 27: 19.
15) Tufarelli C et al. Transcription of antisense RNA leading to gene silencing and methylation as a novel case of human genetic disease. 2003. Nat Genet 34: 157-165.
16) Faghihi MA et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. 2008. Nat Med 14: 723-730.
17) Moseley et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. 2006. Nat Genet 38: 758-769.
18) Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. 2006. Annu Rev Neurosci 29: 259-277.
Regulation of Expression
The question of whether lncRNA is functional has led to careful study of its expression patterns, to determine whether or not they are regulated. If lncRNA transcription is due to random transcriptional noise, the expression levels of lncRNA transcripts would not be expected to vary spacially, temporally, or in response to stimuli (R2,R3). However, differences have been observed in the expression levels lncRNAs between tissue types (10). Some lncRNAs also show very precise expression patterns in tissues. For example, Mercer et al observed patterning of lncRNA expression in the mouse brain both in the tissue as a whole as well as subcellular locations (11). Expression of some lncRNAs has also been demonstrated to be developmentally regulated (12).
Location in Genome
LncRNAs can be categorized according to their proximity to protein coding genes in the genome, using this criteria lncRNAs are generally placed into five categories; sense, antisense, bidirectional, intronic, and intergenic (R2, R3).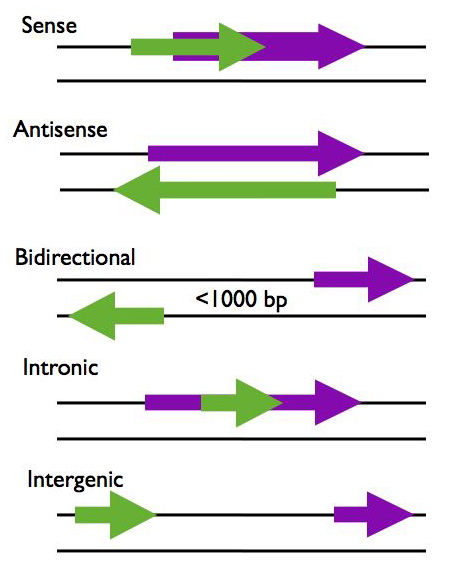
- Sense - The lncRNA sequence overlaps with the sense strand of a protein coding gene.
- Antisense - The lncRNA sequence overlaps with the antisense strand of a protein coding gene.
- Bidirectional - The lncRNA sequence is located on the opposite strand from a protein coding gene whose transcription is initiated less than 1000 base pairs away.
- Intronic - The lncRNA sequence is derived entirely from within an intron of another transcript. This may be either a true independent transcript or a product of pre-mRNA processing
- Intergenic - The lncRNA sequence is not located near any other protein coding loci.
Post Transcription Forms
It is possible that many lncRNA transcripts are not end products, but are further processed into a final functional form. The presence of sense lncRNAs which contain exons from mRNA sequences and intronic lncRNA that are derived entirely from the intronic sequence of mRNAs has lead to the hypothesis that many lncRNA transcripts are unprocessed pre mRNAs that will be spliced to establish their open reading frames and that intronic lncRNAs are byproducts of this splicing (R1,R2,R3). However, this is not the case for all sense and intronic lncRNAs because the expression patterns of some of these transcripts are not the same as their associated protein coding gene, instead there are many examples that have independent expression patterns (11). Another hypothesis suggests that some lncRNA sequences are precursors to short ncRNAs that have defined regulatory functions such as microRNAs.An example of this is the lncRNA H19 which contains the exon that encodes the microRNA miR-675 (R3). Based on this evidence, post-transcriptional processing may occur with many lncRNA transcripts, but until more lncRNAs are functionally defined we will not know what proportions of them are precursors or independently functioning molecules.
Secondary Structure
One method to try to predict lncRNAs that have defined roles in the cell is to use computer models to try and determine which lncRNA sequences form consistent secondary structures such as short stem loops (R3). Secondary structure formation is an important consideration in lncRNAs because they are be able to interact with proteins or genomic DNA via these structures. Recently, scientists have attempted to use models that predict secondary structure to redefine the question of lncRNA evolution by looking at sequence conservation, not of the lncRNA sequence as a whole, but instead at sequence conservation or compensatory mutations that would maintain secondary structure motifs (13). This idea is promising, but is currently hampered by limitations in the sophistication of the computer models available to accurately predict RNA secondary structures.
Some well known example members of this class of RNA are Xist/Tsix, H19, HOTAIR, and Air.
LncRNA expression profiles are altered in in several types of cancers, including human prostate cancer, renal cell carcinomas, breast cancer, ovarian cancer, and human lung adenocarcinomas (R1), raising the possibility lncRNA expression profiling may be an informative biomarker in cancer diagnoses (14).
Long noncoding RNA is abbreviated as lncRNA.
Introduction
Long noncoding RNAs (lncRNA) are transcribed RNA molecules greater than 200 nucleotides in length. The existence of individual lncRNAs such as H19 and Xist has been known since the 1980s; these lncRNAs were discovered using traditional gene mapping approaches and were only discovered to be non protein-coding after their RNA sequences were analyzed (1,2). LncRNAs were generally considered to be anomalies until the advent of technologies that were capable of unbiased high throughput sequencing of all the expressed transcripts in cells. The extent of lncRNA transcription was first revealed in a study using large-scale sequencing of cDNA libraries in the mouse (3). This study by Okazaki et al determined that a large proportion of the mammalian transcriptome does not code for proteins and defined lncRNAs as a significant class of transcripts. The more recent development of sensitive tiling microarrays has only increased the numbers of known lncRNAs, currently it is known that lncRNA transcripts far exceed the number of protein coding mRNAs in the mammalian transcriptome (4). For example, the mouse transcriptome was recently calculated to contain approximately 180,000 transcripts organized into 44,000 transcription clusters however only approximately 20,000 of these transcripts are thought to be protein coding genes (4,5).
Protein genes vs RNA genes: Numbers and Implications
Since the completion of the Human Genome Project our perception of our genome has undergone a dramatic shift. The number of protein coding genes in our genome has been revised downward multiple times, whereas the number of known non protein coding transcripts has increased exponentially over the past decade (4,5). Our improved understanding of the content of the mammalian transcriptome has raised questions about the the classical definition of a “gene” as a genetic sequence that codes for protein and has enormous implications for future genetics research.
Future Studies
LncRNA has been described as part of the “dark matter of the genome”, due to the fact that although we are now able to detect its presence, its function and activity remains poorly understood. Whether or not lncRNA transcription is functionally significant remains a fundamental question in this field of study and future studies of lncRNA are needed to fully understand its role in our genome.
In addition to the general alterations in lncRNA expression profiles in cancer states, specific lncRNAs have also been shown to be involved in a variety of human diseases (R2,R3). Some examples are described below.
Alpha-Thalassaemia
An inherited form of alpha-thalassaemia is caused by the translocation of of an antisense lncRNA to a location near the alpha-globin gene (HBA2). The translocation and induction of expression of this lncRNA results in the epigenetic silencing of the HBA2 gene and results in this form of human anemia (15).
Alzheimer’s Disease
The beta-secretase-1 (BACE1) protein is a crucial enzyme in the progression of Alzheimer’s disease. In a recent study, the increased expression of the antisense transcript of the BACE1 gene in response to cell stressors such as amyloid-beta 1-42 has been implicated in the progression of Alzheimer’s disease (16).
Human Spinocerebellar Ataxia Type 8 (SCA8)
Patients with SCA8 have been shown to have a trinucleotide expansion in an lncRNA named ataxin 8 opposite strand (ATXN8OS) which is antisense to the KLHL1 gene. The involvement of this mutation in SCA8 disease progression has been confirmed in a transgenic mouse model. Transgenic mice with this repeat expansion show a similar progressive neurological phenotype to humans with SCA8 (17).
Myotonic Dystrophies
Repeat expansions in lncRNAs are also involved in multiple organ system myotonic dystrophies. These expansions in lncRNA sequences are predicted to alter the lncRNA secondary structures such that they prevent splicing regulators from interacting normally with pre-mRNAs (18).